| Amyloidose |
| |
|
| |
HER 97 / dRob 210-220 / Laissue 248-249, 312 |
| |
|
| |
|
| |
|
| |
| •
|
|
bindegewebige und perivaskuläre
Ablagerung von fibrillären (b-Faltblatt)
Proteinen ("Amyloid") und nachfolgende
Störung des Stoffaustausches, Druckatrophie oder
mech. Behinderung der Kontraktion von Myokard-
und glatten Muskelzellen. |
| • |
|
Amyloid
ist für proteolytische Enzyme nicht verdaulich,
einmal angelegte Depots bleiben bestehen. |
| • |
|
kleinere
Depots haben keinen Effekt; Wrk erst durch
excessive Ablagerungen. |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| •
|
|
AA |
=
AkutphasenProtein (Amyloid AA) |
|
ï |
|
bei
chron. Entzündungen |
|
|
|
| •
|
|
AL |
=
leichte Ketten (Lambda-Ketten, Amyloid AL) |
|
ï |
|
z.B.
B-ZellTu, va MultiplesMyelom |
|
|
|
| |
|
|
|
|
ð |
|
AK
gg. leichte Ketten binden Amyloid-AL nicht mehr! |
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ð |
|
|
insbesondere bei Herzbeteiligung
wg zunehmender MyokardInsuffizienz ungünstig |
| ð |
|
|
lokal umschriebene Amyloidosen
sind idR harmlos |
|
| |
|
| |
| Einteilung nach dem
beteiligten Protein |
|
| |
| Amyloidtyp |
Vorläuferprotein
im Serum
|
Klinisches
Syndrom
|
AL
A-Kappa
A-Lambda |
monoklonale
leichte Ig-Ketten (Lambda od. Kappa) |
B-Zell-Tu
wie multiples Myelom (5-11%), Bence-Jones-Plasmozytom,
M. Waldenström, benigne (monoklonale)
Gammopathie; entspricht der früheren Bez. primäre
Amyloidose. Ablagerung va im Herzen,
GIT (RektumBx meist positiv),
Haut und SkelettMuskel. Aber ebenfalls wie AA in
parenchymatösen visz. Organen (Leber, Milz,
Niere, NN).
|
| AA |
Serum-Amyloid-A |
Amyloidose
reaktiv bei chron. Entzündungen; heute va RheumatoideArthritis
(5-12%), Spondylitis Ankylosans, Colitis
Ulcerosa, Osteomyelitis, Lepra, fam.
Mittelmeerfieber oder Tumoren (Hypernehprom, M.
Hodgkin); früher va nach Tbc. entsprichtder früheren
Bez. sekundäre Amyloidose, idiopathische
Amyloidose vom AA-Typ
Ablagerung va in
parenchymatösen visz. Organen (Leber,
Milz, Niere, NN)
|
| AB |
Beta-2-Mikroglobulin |
Dialyse-Arthropathie |
| AFp |
Präalbumin-Homolog |
hereditäre
Amyloidose bei portugiesischen fam.
Polyneuropathien |
| AEt |
Calcitonin-Homolog |
idiopathisch
bei medullärem Schilddrüsen-Ca |
| ASc1 |
Präalbumin-Homolog |
idiopathische
senile Herzamyloidose |
| ASb |
unbekannt |
idiuopathische
senile und präsenile GehirnAmyloidose |
| unbekannt |
unbekannt |
Haut-,
Corneaamyloid, Knorpelamyloid u.a. |
|
| |
|
| |
|
| |
| |
| ð |
|
fortschreitender
Funktionsverlust der betroffenen Organe |
|
|
|
|
| |
|
(Leber,
Herz, Niere, KM, GIT, Atemwege, Haut) |
|
|
|
|
| |
|
|
|
|
|
|
| •
|
|
u.a.
Polyneuropathien |
|
|
|
|
| •
|
|
u.a.
Karpaltunnelsyndrom |
|
|
|
|
| •
|
|
u.a.
arthritische Beschwerden |
|
|
|
|
| •
|
|
u.a.
diastolische Dysfunktion, HerzrhythmusStörungen |
|
ð |
|
zunehmende
Myokardinsuffizienz |
| •
|
|
u.a.
nephrotisches Syndrom, terminale NI |
|
|
|
|
|
| |
|
 |
| • |
|
funktionelle
einschränkungen siehe oben |
|
|
|
|
| • |
|
ev.
Makroglossie |
|
ï |
|
bei 20% der AL |
| • |
|
Hautveränderungen |
|
|
|
|
| |
|
|
|
|
|
|
| • |
|
Ödematöse
Extremitäten |
|
ï |
|
CardioMyopathie |
| • |
|
Dyspnoe |
|
|
|
|
| • |
|
HerzrhythmusStörungen |
|
|
|
|
| • |
|
Müdigkeit |
|
|
|
|
| • |
|
Gewichtsverlust |
|
|
|
|
| |
|
|
|
|
|
|
| • |
|
schwacher
Handgriff |
|
ï |
|
Einlagerung in
Sehnen und andere Weichteile, PNP |
| • |
|
Schluckschwierigkeiten |
|
ï |
|
sensomot. PNP |
| • |
|
Taubheitsgefühl
in den Gliedmassen |
|
|
|
|
|
| |
|
| |
|
| |
|
| |
 |
| •
|
|
typische
BiopsieOrte: |
|
• |
|
RektumSchleimhaut |
|
ð |
|
Trefferquote
ca. |
|
85% |
|
| |
|
|
|
• |
|
Retinaculum
Flexorum |
|
ð |
|
|
|
100% |
|
| |
|
|
|
• |
|
KM |
|
ð |
|
|
|
40% |
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
| •
|
|
zus.
bei erkennbarem Organbefall |
|
ð |
|
Dünndarm, Leber, Herzmuskel, N.Suralis,
Lippe, Zunge, Niere, Muskel, Haut |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
 |
| •
|
|
ev. abnormes Protein (VorläuferProtein) |
| |
|
|
|
|
|
| |
|
ð |
|
Akute-Phasen-Protein
(Serum-Amyloid A "AA") |
|
| |
|
ð |
|
Lambda-Ketten
von Ig (AL-Protein) |
|
|
| |
|
| |
|
| |
|
 |
ð Robbins 210-220 / Curran 5.10, 10.15-16, 1.38, 8.28
/ Laissue 248-249, 312 |
| |
| |
| •
|
|
Allgemein: |
|
bindegewebige
und perivaskuläre Ablagerung von fibrillären (b-Faltblatt)
Proteinen ("Amyloid") und nachfolgende
Störung des Stoffaustausches, Druckatrophie oder
mechanische Behinderung z.B. der Herzkontraktur.
- allerdings erst bei extensiver Ablagerung.
Kleine Depots haben keine Wirkung. |
| |
|
|
|
|
| •
|
|
Makro:
|
|
speckig-glänzende
OF, transparente Erscheinung |
| |
|
|
|
betroffene
Organe vergrössert, konsistenzvermehrt, hart. |
| |
|
|
|
Makroskopisch
kann das Gewebe mit Lugol-Iodlösung eingefärbt
werden, welches das Gewebe sehr stark anfärbt (wie
pflanzliche Stärke) |
| |
|
|
|
|
| •
|
|
Mikro:
|
|
typisches
Färbeverhalten: Kongorot und grünliche
Doppelbrechung im polarisierten Licht. Für den
Nachweis kleinerer Ablagerungen werden immunolog.
Techniken benutzt. Im HE-Präparat homogenes, stark
lichtbrechendes Material um Gefässe,
Retikulinfasern und BM.
|
| |
|
|
|
|
| •
|
|
DD: |
|
|
|
| |
|
| |
| autoptisch |
| |
|
|
| |
|
|
| quelle |
quelle |
|
| histopathologisch |
 |
 |
 |
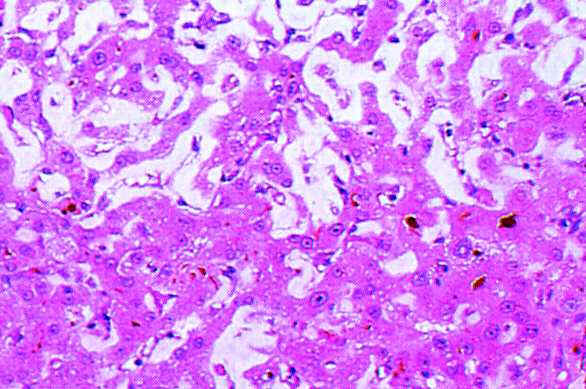
| Amyloidose
der Leber |
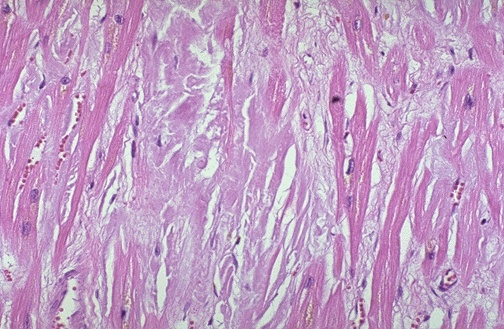
Amyloidose
Herz |
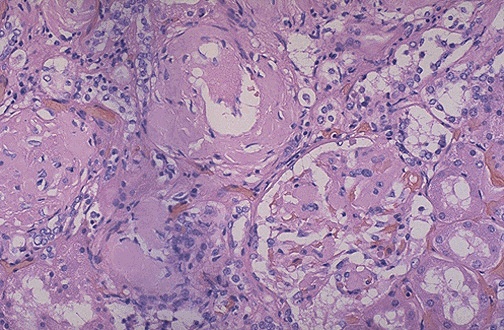
Amyloidose
der Niere |
| quelle
Pschy |
quelle
Utah |
quelle
UniNecker / F |
 |
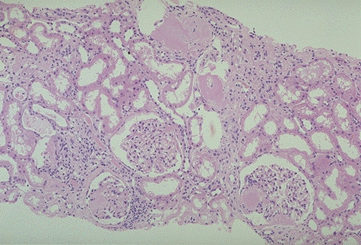 |
 |
Amyloidose der Niere:
Glomeruli mit Amyloid aufgefüllt
Tubuli mit Amyloid-Gurt drum herum und ev. mit
filtriertem Protein verstopft. |
gleiches
Bild in Kongorot |
| quelle
UniNecker / F |
quelle
Jichi/Jap |
quelle
Jichi/Jap |
|
| |
|
 |
| Praep |
Bemerkung |
| 21.3 |
|
Makro:
bei extensiver Einlagerung Splenomegalie
bis 1kg (no: 130-150g) möglich.
Wachsartige, leicht transparente
Erscheinung.
zwei verschiedene makroskopische
Verteilungsmuster:
• Sago-Milz: Malpighi-Körperchen
(Milzfollikel, := weisse Pulpa)
• Schinken-Milz:
primär rote Pulpa betroffen. |
| Mikro:
In den Arteriolenwänden eingelagert und
entlang der Sinuslichtungen ein sich
homogen färbendes Material. Einige Gefässe
sind obliteriert. Vermutlich Milz vom
Schinkentyp. |
| Lit:
(Milz: BucherWartenberg 224) |
|
| 54.1 |
|
| Makro:
Festes und elastisches Organ, das
Parenchym erscheint blass, wachsartig und
etwas transparent. |
Mikro:
Die ersten Ablagerungen
werden im Disse-Raum angelegt. Mit
zunehmender Menge kommt es zur
Druckatrophie der Leberzellplatten. Das
Leberparenchym kannzu grossen Teilen
durch Amyloid ersetzt sein, denoch kommt
es selten zur schweren Leberfunktionsstörung.
Übersicht:
homogenes, kongorotes Material, das ungefähr
die Hälfte der Schnittfläche einnimmt.
Die Leberzellplatten sind in einigen
Regionen vollständig verschwunden.
Vergrösserung:
(sehr fortgeschrittenes Stadium!)
zwischen den massiven Amyloidablagerungen
"eingeklemmte"
Leberzellplatten; in einigen Zonen sehr
druckatrophisch. Bei diesem Aspekt ist
die Tatsache einer nur leichten
Leberinsuffizienz erstaunlich! |
| Lit:
Robbins 210-220 / Laissue 248-249, 312 /
curran 5.10, 110.15-16, 1.38, 8.28 |
|
| 54.2 |
|
| Makro:
Potentiell für den
Patienten die gefährlichste
Organmanifestation. Die Niere kann normal
erscheinen oder aber von fester Konsistez
und vergrössert, blass grau und
wachsartig transparent sein. Im
Endstadium kann die Niere schrumpfen, z.T.
wegen der vaskulären Verengung durch die
Amyloiddepots. |
Mikro:
Die Ablagerungen finden sich in
der Basalmembran der Tubuli, Arteriolen
und glomerulären Kapillaren. später
obliteration der Glomerulusschlingen und
tubuläre Atrophie. Durch den Verlust von
Podozytenfüssen und durch die
Ablagerungen in der BM kommt es zum
Vberlust der selektiven Filterfunktio und
zur Proteinurie, die bis zum
nephrotischen Syndrom fortschreiten kann.
später nimmt infolge Gefässobliteration
die Primärharnmenge ab.
Übersicht:
deutliche Vermehrung des
Interstitiums zwischen den Tubuli, einige
mit kongorotem Material angefüllte
Glomeruli.
Vergrösserung:
Einzelne Tubuli
sind von einem deutlichen hyalinen kongoroten
Band umgeben. In einigen
befindet sich ein scholliges,
proteinhaltiges Material. Die Glomeruli
sind durch die Zunahme des
mesangialen Raumes erweitert (Nur
noch Amyloid"), so dass es zum
Obliteration des Harnraumes kommt. Die
meisten Kapillarlumina er Glomeruli sind
verschlossen. Deutlich sind die
Amyloidablagerungen in den Arterien zu
erkennen.
"Tubuli: Gurt aus Amyloid
und filtrierte Proteine im Lumen /
Glemeruli: mit Amyloid aufgefüllt" |
|
| 54.3 |
|
| Allgemein:
Die Depots beginnen um die
Gefässbasalmembranen des Kortex,
normalerweise in der Zona glomerulosa (äusserste
Schicht, Mineralokortikoide). später
progredieren die Ablagerungen in tiefere
Zonen. Druckatrophie. Dieser Prozess kann
bis zur Nebenniereninsuffizienz
fortschreiten. |
Übersicht:
In diesen Präparaten sind die
Amyloidablagerungen nicht nach Lehrbuch
von aussen nac innen fortschreitend. Sie
sind hier am stärksten in der Zona
fasciculata (Glucocorticoide) deren Säulenform
durch das Amyloid noch verstärkt wird.
Vergrösserung: starke
autolytische Veränderungen. |
| Lit:
BucherWartenberg: NNR: 322 |
|
| 54.4 |
|
| Makro:
siehe 21.3 |
Mikro:
Die kongoroten Amyloidablagerungen
befinden sich va in der ws Pulpa um die
Zentralarterien = SagoTyp
Vergrösseruing:
Das follikuläre Lymphgewebe (Malpighi-Körperchen)
ist weitgehend durch Amyloid ersetzt.
Deutliche Amyloideinlagerungen auch in
den Arterien. |
|
|
| |
|
| |
|
 |
|
| |
|
|
| •
|
|
idR
wenig erfolgversprechend |
|
|
| •
|
|
organbezogen
symptomatisch |
|
|
| |
|
|
|
|
| • |
|
umschriebene
Amyloidablagerungen bedürfen idR keiner Therapie |
|
|
| |
|
|
|
|
| ð |
AA |
|
| |
|
Grundkrankheit
behandeln |
|
|
| |
|
ev.
Mobilisierung der abgelagerten Amyloidmassen mit
Dimethylsulfoxid versuchen |
z.B.
mg/d |
•
K: |
| ð |
bei fam. Mittelmeerfieber (ebenf.
AA) |
|
| |
|
lebenslange
Gabe von Colchizin |
z.B.
mg/d |
•
K: |
| |
|
oder NSAR |
z.B. mg/d |
• K: |
| |
|
Steroide
sind wirkungslos ! |
|
|
| |
|
|
|
|
| ð |
AL |
|
| |
|
zytostatische
Behandlung des B-Zell-Tu |
|
|
| |
|
|
|
|
|
| |
|
| |
|
| |
|